Introduction
The specific detection of pathogenic microbes is a useful application of high-throughput genomic sequencing that has generated substantial interest in the clinical, public health, and biodefense communities. Rather than focusing on a limited set of biochemical signatures, such as a surface antigen or conserved marker gene, whole genome analysis offers the potential for highly sensitive and accurate detection that stands up robustly in the face of trace-level input, unculturable target microbes, and genomic sequence divergence. However, existing tools for microbial metagenomics are typically focused on identifying the broadest possible range of organisms, potentially sacrificing computational or biological specificity for taxonomic breadth. This can often lead to several problems, including false positives, results that are more accurate at the genus level than the species level, or taxonomic assignment that uses some parts of the “core genome” but that neglect the unique biological and virulence markers of a species. This issue was recently highlighted in the case of the putative pathogen identification (B. anthracis) in two samples in the New York City subway system
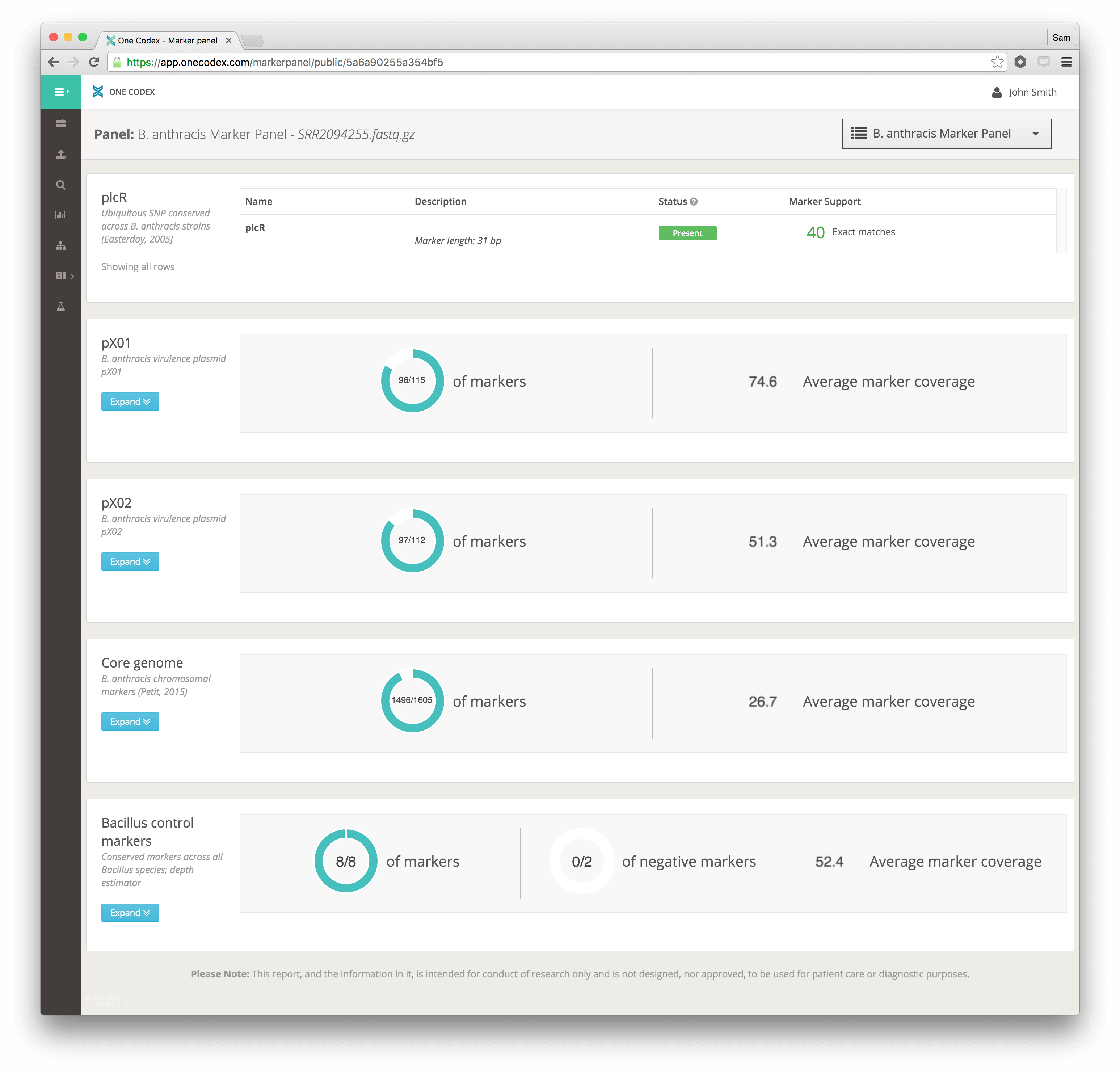
To complement these broad-based metagenomic methods, we constructed a targeted bioinformatic tool to detect Bacillus anthracis that takes into account the specific biology of the pathogen, including the virulence plasmids pXO1 and pXO2 and the plcR SNP that has been previously identified as being diagnostic of B. anthracis. The resulting “marker panel” on the One Codex platform provides applied microbiologists with a straightforward and powerful tool for the detection of B. anthracis, and we believe that it also demonstrates a generalizable approach for the development of easy-to-interpret targeted assays that complement exploratory metagenomic analysis. Figure 1 provides a visual summary of the highly sensitive detection provided by this panel, while Figure 2 shows the interface for an individual result. The design, construction, and evaluation of the panel is elaborated further in the sections that follow.
Methods
Marker Panel Construction
A “Marker Panel” on the One Codex platform consists of a set of target sequences that are compared against an input dataset of short reads, long reads, assemblies, or complete genomes (in either FASTA or FASTQ format). A marker panel summarizes samples by the aggregate coverage, depth, and nucleotide identity of all input sequences aligned against the carefully selected target sequences. These target sequences are grouped for ease of interpretation and presented to the end user with group-level summary statistics. An example Marker Panel result page is shown in Figure 2.
The B. anthracis marker panel was assembled to detect the presence of B. anthracis using four specific types of probes:
- plcR SNP: A single SNP was identified by Easterday, et al. as being ubiquitous and specific to B. anthracis.
A 31bp marker spanning that SNP was used as a target sequence and is displayed in the top panel of Figure 2. - Virulence plasmid markers: The complete set of 50bp sequences from the pXO1 and pXO2 plasmids were compared against the One Codex Database (containing roughly 40,000 complete microbial genomes), and all non-overlapping markers specific to B. anthracis were used as plasmid-specific target sequences (pXO1 – n=115; pXO2 – n=112), displayed in the second and third panels in Figure 2.
- Core genome SNPs: A set of 31bp k-mers were identified by Petit et al. as being specific to the main chromosome of B. anthracis
, and not shared by any other Bacillus species. A non-overlapping set of 1,605 of these target sequences was used for the “Core Genome” panel shown in the fourth panel of Figure 2. - “Near neighbor control markers”: A final panel presents select highly conserved markers present across Bacillus species. These markers do not indicate the presence of B. anthracis, but rather serve to estimate the relative depth of sequencing across all Bacillus species present in a sample. The resulting coverage statistics can also be used to distinguish between the genuine but low-level presence of B. anthracis and sequencing error in a deeply sequenced sample.
B. anthracis Marker Panel on One Codex
The B. anthracis detection panel can be used on the One Codex platform by uploading FASTQ datasets via the GUI interface or the command line client (details here). An example of the results page is shown in Figure 2. Please contact the authors for further guidance on the use of this tool on One Codex.
Marker Panel Execution
The B. anthracis detection panel is executed on the One Codex platform as part of an automated analysis. Each sequence in an input dataset is compared against every target sequence with a minimal exact match length of 31bp and a tolerance of up to 3 SNPs. For those markers 31bp in length, an exact match is required for detection. The platform records the number of input sequences matching each target, and presents the user with a single display that summarizes the abundance of these 1,841 markers across four categories (plcR, pXO1/pXO2, the main chromosome, and the cross-Bacillus depth controls). An example of this display is shown in Figure 2.
Inclusivity & Exclusivity Testing
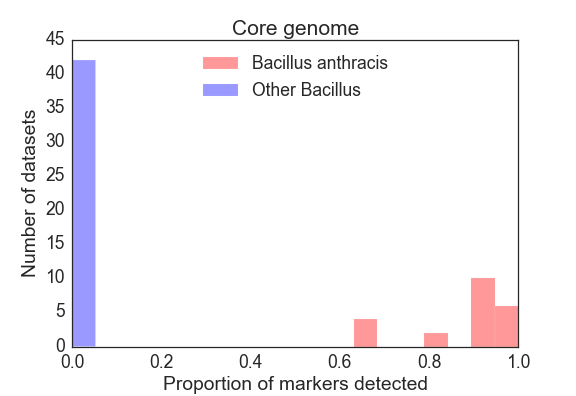
The accuracy of the B. anthracis marker panel was assessed by analyzing a diverse collection of B. anthracis isolates available from public repositories as short-read shotgun sequencing datasets. A minimum threshold of 3X average sequencing depth was used to ensure sufficient genome coverage, and datasets were all downsampled to <=20X genome coverage to control for the contribution of low-frequency sequencing error. For each of those input datasets, we quantified the number of markers detected from each category. Those results were compared against those from a collection of non-B. anthracis isolates from the genus Bacillus that were also available from public repositories. The number of datasets analyzed from each species is shown in Table 1, and the complete list of datasets is available as Table S1. Table 2 includes links to 3 example analyses. The results of this analysis are shown in Figure 3.
| Species | Number of samples |
|---|---|
| Bacillus anthracis | 22 |
| Bacillus thuringiensis | 30 |
| Bacillus cereus | 9 |
| Bacillus mycoides | 2 |
| Bacillus subtilis | 1 |
| Name | Description | B. anthracis markers detected / total | B. anthracis depth / Bacillus depth |
|---|---|---|---|
| SRR2175367 | WGS of Bacillus anthracis isolate BA1015 (Los Alamos National Laboratory) | 1527/1605 | 0.51622 |
| NYC Subway - Pelham Pkwy | Metagenomic sequencing of NYC Subway - 4.5M reads | 1/1605 | 0.00009 |
| NYC Subway - J Train | Metagenomic sequencing of NYC Subway - 3.4M reads | 4/1605 | 0.00008 |
Limit of Detection Analysis
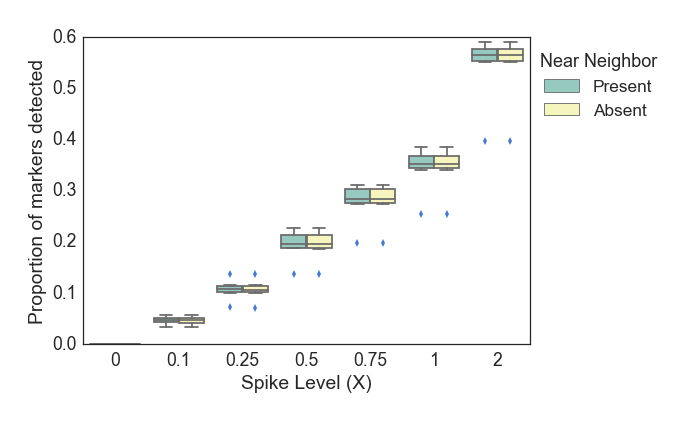
We executed the detection panel using in silico mixed samples with target organisms present at a range of abundances. The background for all of the in silico spike-in samples were 5 million reads from a soil sample (SRR351473) that was sequenced as part of the Great Prairie Soil Metagenome project (PRJNA50473). Six WGS datasets were spiked in at a range of abundances (n=6) from 0.1X to 2X sequencing depth. A second series of samples was created that also included 150K reads from a near neighbor (B. thuringiensis), for roughly 8X sequencing depth. For each of these datasets, the proportion of markers detected was calculated and plotted as a function of the spike-in level and near neighbor presence Figure 6.
Results
Inclusivity & Exclusivity Testing
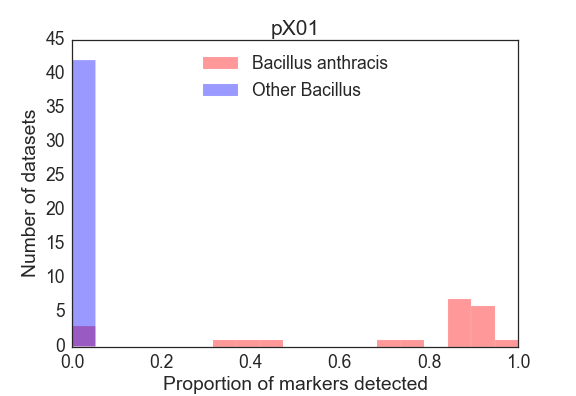
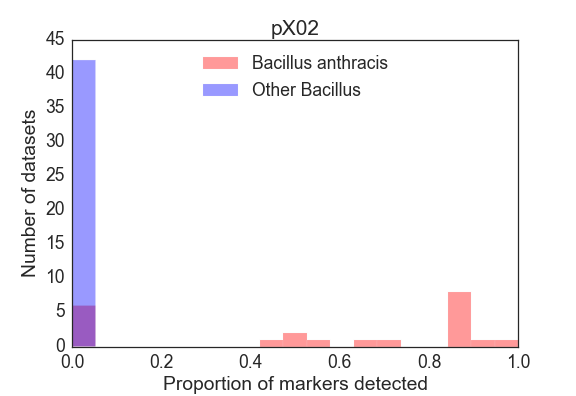
For each sample, we summarized the result of the detection panel as the proportion of markers detected from the main chromosome, as well as for the plcR SNP and pXO1 and pXO2 plasmids. For the core genome, Figure 3 shows a frequency histogram of the proportion of markers detected for the target organism (B. anthracis) and other Bacillus speices. The lowest proportion of core genome markers detected in a B. anthracis dataset was 65.2%, and the highest proportion of core genome markers detected in the exclusion dataset was 2.5% (for SRR2157174). The plcR SNP was detected in 100% of all B. anthracis isolates and in none of the other Bacillus isolates. The pXO1/pXO2 virulence plasmids were found to varying degrees in these B. anthracis samples (Figure 4 and Figure 5), consistent with both genomic heterogeneity in these mobile elements, as well as a number of B. anthracis isolates in the public repositories that entirely lack those plasmids.
Limit of Detection Analysis
Analysis of in silico mixed samples containing B. anthracis or other Bacillus at a range of low abundances is shown in Figure 6. The proportion of detected markers increases roughly linearly with the amount of target genome coverage in the input dataset. The presence of a near neighbor (B. thuringiensis) at a high abundance (~10X), does not impact the proportion of B. anthracis markers detected. The plcR SNP was detected in 33.3% of samples with a spike-in level of 0.1X and above, 50% of samples at 0.75X, and 83.3% of samples at 2X spike-in. This rate of detection is consistent with the expected distribution of random sampling across the core genome. As Figure 6 demonstrates, these detection rates are robust to the presence of a near neighbor, even when the spiked in B. thuringiensis is ~100X more abundant than the target B. anthracis. Figure 7 shows the detection of B. anthracis scaled to the total abundance of Bacillus species in these synthetic spike-in samples. These data show that even at 0.1X spike-in, when the near neighbor is present in ~100-fold excess of B. anthracis, the proportion of estimated sequencing depth for B. anthracis compared to all Bacillus ranges from 0.005-0.008, substantially above the level expected due to sequencing error.
Discussion
The extreme virulence and environmental stability of Bacillus anthracis requires high-confidence assays that simultaneously enable trace detection and are robust to reporting alarming false positives. Moreover, the specific biology of B. anthracis and other Bacillus species requires an assay that independently characterizes both the core genome and virulence plasmids in the context of complex metagenomic samples. Indeed, Bacillus cereus isolates have been recently found containing the pXO1 and pXO2 virulence plasmids and causing Anthrax-like disease.
High throughput sequencing offers a promising foundation for such an assay, but the inherent scale and complexity of the resulting datasets tends to confound “one size fits all” analytic approaches (doubly so for metagenomic samples). Indeed, this inquiry was motivated by an alarming false positive arising from the broad application of exploratory metagenomic tools.
Here, we present a targeted assay for the detection of Bacillus anthracis which is specifically designed with the biology of the pathogen in mind and complements, rather than replaces, unbiased metagenomic analysis. Initiated automatically or at the behest of the end user, the assay detects the diagnostic plcR SNP, the pXO1/pXO2 virulence plasmids, the core B. anthracis genome, and a set of sequences conserved broadly across Bacillus species.
We tested the detection panel against a diverse set of previously-sequenced Bacillus species – demonstrating its sensitivity across a range of B. anthracis strains, sequencing platforms, sequencing depths, and the presence or absence of confounding near neighbor species. Indeed, all of the 23 analyzed B. anthracis isolates contained 65.2% or more of the core genome markers, while all of the 42 analyzed other Bacillus isolates contained 2.5% or lower core genome markers (Figure 3). Moreover, the limit-of-detection analysis shows that this assay was extremely robust to the presence of a near neighbor Bacillus (Figure 4).
This assay directly addresses the potentially confounding scenario in which a small number of B. anthracis SNPs are present in a deeply-sequenced metagenomic sample. Those sequences may be present due to trace levels of the pathogen, but they may alternately be caused by a large number of reads from another Bacillus species and a low level of sequencing instrument error. To distinguish between these possibilities, we present the level of evidence for B. anthracis in the context of the relative abndance of all Bacillus species in a sample. This allows an end user to directly compare the relative abundance of B. anthracis to other Bacillus species and more accurately judge whether a given sample merits concern. We also believe that this panel offers a model for the development of easy-to-interpret sequencing-backed assays that serve the needs of applied end users.